






An genetically determined process that results in plasma membrane rupture and the release of intracellular content, so-called damage associated molecular patterns (DAMPs), is defined as Regulated Necrosis. Pathways of Regulated Necrosis include but are not limited to necroptosis and ferroptosis. We demonstrated that necroptosis and ferroptosis are independent signaling pathways that contribute to the organ damage. We therefore believe that targeting both pathways with combinatory therapies may be the most effective treatment.
Necroptosis
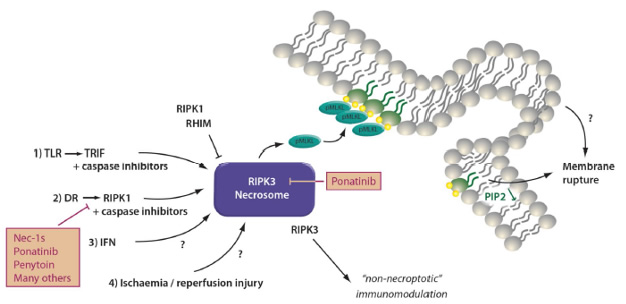
The signaling pathway of necroptosis: Phosphorylated mixed linage kinase domain like (pMLKL) is the only known mediator of necroptosis and its detection defines the activation of this pathway. Potentially, several kinases may phosphorylate MLKL, but only receptor interacting protein kinase 3 (RIPK3) has been described while this review was written. MLKL carries several phosphorylation sites, and RIPK3 phosphorylates the so-called “activation loop”. However, complete function of pMLKL requires the depohsphorylation at the hinge region that unleashes the deadly activity of a 4 helical bundle (4-HB) which binds PIP2 in the plasma membrane and tends to oligomerize with other pMLKL molecules. Downstream of pMLKL, sensitivity of cells to undergo necroptosis is controlled by proteins that control membrane blebbing and microvesical formation. Unlike previously suggested, pMLKL does not directly form pores in the plasma membrane of cells. Full activation of RIPK3 requires the assembly of the necrosome, a higher order structure that consists of oligomerized RIPK3 molecules that are stabilized by HSP90 and CDC37, two chaperones the loss of which results in defective necroptosis. Several triggers result in the formation of the necrosome. Death receptor (DR), e.g. TNFR1-stimulation in the presence of a caspase-inhibitor or a dysfunctional caspase-8 represents the most prominent and best investigated stimulus that requires RIPK1 kinase activity for necrosome formation. RIPK1, as RIPK3, contains a rip homotypic interacting motif (RHIM)-domain that intercalates with the RHIM of RIPK3 and prevents necrosome assembly. Inhibitors of RIPK1 kinase activity, such as Nec-1s and ponatinib, maintain the inactive state of RIPK3 potentially by keeping RHIM-RHIM-interactions intact. Necrosome assembly has been repeatedly reported downstream of Toll like receptors that bind to the intracellular adapter protein TRIF which also contains a RHIM domain and activation of this pathway results in robust RIPK1-independent necrosome formation. In vivo, reperfusion following ischemic injury severely triggers necroptosis, and several other models have been described, such as injection of recombinant human TNF-α into mice. In addition, protein kinase R (PKR) and DAI, a protein that is capable of but functionally not limited to viral DNA sensing activation as triggered by interferons, can activate RIPK1-mediated necrosome formation, but the relative contribution of these two factors remains unclear. Certainly, DAI robustly triggeres necrosome formation via its RHIM domain, possibly via nuclear signalling. However, importantly, necroptosis contributes to acute kidney injury in some models, such as ischemia, but inhibition of necroptosis does not affect other models, such as foliac acid induced AKI.
Ferroptosis
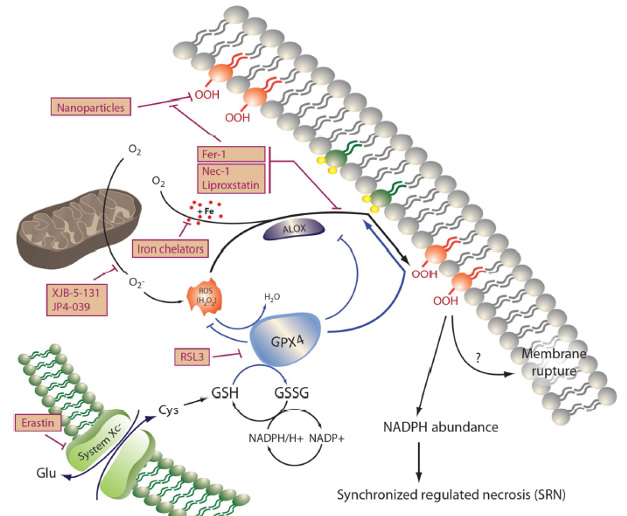
The signaling pathway of ferroptosis: Peroxidation of membrane lipids, predominantly phosphatidylinositole and phosphotidylethanolamine, represents the point of no return during ferroptosis that results in loss of NADPH-abundance and synchronized regulated necrosis (SRN) in sensitive organs, such as the renal tubular compartment or the myocardium. Lipoxygenases (ALOX) mediate lipid peroxidation and are kept in their inactive state by the constitutively active function of glutathione peroxidase 4 (GPX4), a selenoenzyme that requires glutathione (GSH) to function. Ferroptosis may be triggered by inhibition of system Xc-minus, a cys/glu-antiporter in the plasma membrane by a lethal compound referred to as erastin. Inhibition of system Xc-minus functionally inhibits the activation of the GSH-synthase (GSSG), resulting in GSH depletion, dysfunction of GPX4 and ferroptosis. RSL3 induces ferroptosis directly by inhibition of GPX4, and certain nanoparticles are capable of inducing ALOX activation in vitro. Before lipid peroxidation occurs, it may be prevented by the ferrostatins liproxstatin, ferrostatin-1 (Fer-1), necrostatin-1 (Nec-1), and the novel compounds 16-86, XJB-5-131 and JP4-039. The latter carries intrinsic anti-necroptotic activity beyond its function as a ferrostatin. Ferroptosis accounts for the majority of tubular cell loss during acute kidney injury because of its mechanism of SRN that shuts down an entire functional unit, but ferroptosis may be triggered by any cell death that occurs in cells of the functional syncytium because of the preterminal loss of NADPH that occurs in all cell death pathways.